This column is the second in a four-part series from Taeho Jo, PhD, assistant professor of radiology and imaging sciences at the Indiana University School of Medicine, titled "AI in Medicine: From Nobel Discoveries to Clinical Frontiers."
Read Column 1: AI Innovation Through the Lens of the 2024 Nobel Prizes
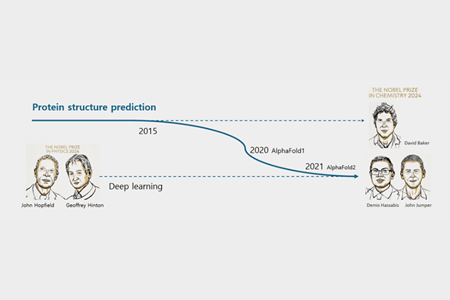
The area where these two utilized AI was protein structure prediction. They, along with David Baker, PhD, who had been researching this field for a long time and also adopted AI later, were awarded the 2024 Nobel Prize in Chemistry.
So, what is protein structure prediction, and why is it important?
The process of creating balloon art provides a useful analogy. A single long balloon can be twisted into a dog or a flower. For this long balloon to become a 3D creation, it must be folded according to specific rules. Depending on which part is folded, the direction of the fold, and the method used, the simple 1D balloon transforms into a remarkable 3D shape.
Proteins in our bodies are very similar. Amino acids, created based on DNA instructions, are linked together in a long chain. Then, they fold in a specific manner to form a 3D protein structure. These resulting proteins are essential components that compose our bodies and regulate life processes.

Predicting the 3D structure of these proteins poses a fundamental challenge in the life sciences. All life processes in our bodies occur through proteins, and for proteins to function correctly, they must have the accurate 3D structure. When a protein folds incorrectly, diseases such as Alzheimer's or Parkinson's can develop. Conversely, understanding the precise structure of a protein can significantly enhance drug development and research into protein function.
There was a significant mystery. We knew the sequence of amino acids and could observe the final shape of the protein, but we didn't fully understand the folding process itself—the rules governing how it folded. It's like having a long balloon and the finished 3D balloon artwork, but not knowing how to fold it.
To address this issue, the scientific community established a competition called CASP (Critical Assessment of protein Structure Prediction). Launched in 1994, this competition takes place every two years, bringing together the world's leading researchers to demonstrate their protein structure prediction skills.
Here's how the competition works: The organizers provide the amino acid sequence of a protein whose structure has just been experimentally determined but has not yet been publicly released. Participants must predict the final 3D structure using only this sequence. Using the balloon analogy, it's like being told only the length and material of the balloon and being asked, "Predict what shape this will fold into."
For instance, consider the rankings from the 11th CASP competition in 2014.

The top-ranked team at that time was Baker's team, one of this year's Nobel laureates.
The second-place team was led by Jianlin Cheng, PhD, from the University of Missouri, with which I was affiliated. In this competition, Cheng's team first introduced deep learning for protein structure prediction (Jo et al., 2015). Back in 2014, there wasn't much data accumulated, and algorithms and hardware for deep learning weren't as advanced as they are today; as a result, we didn't achieve first place. So, how accurate were the protein structure predictions at that time?
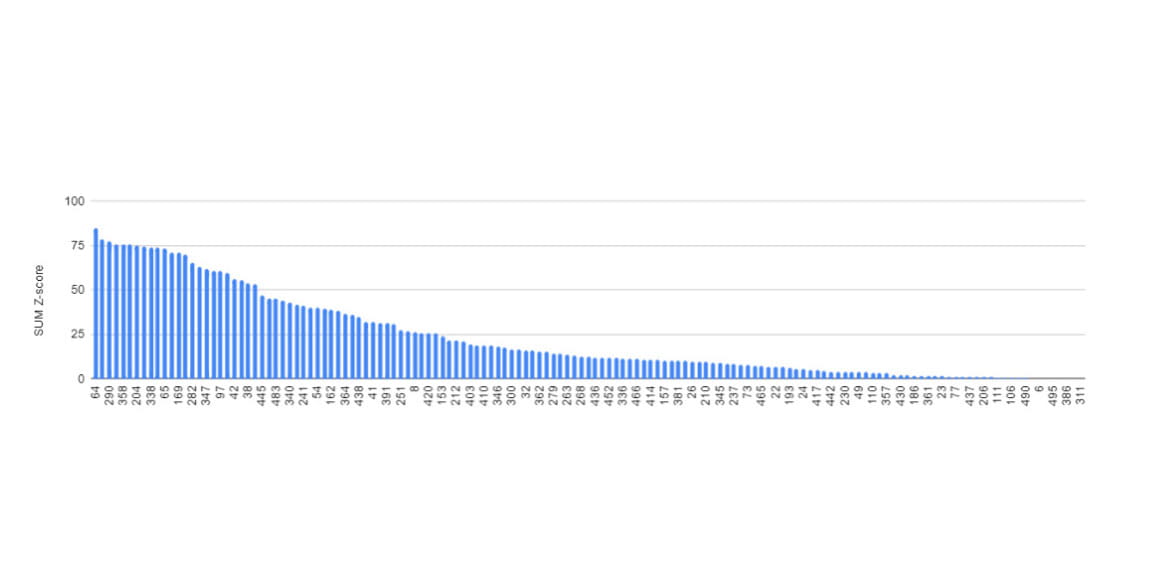
This graph illustrates the prediction accuracy of each research team. The higher the value on the vertical axis (SUM Z-score), the more accurate the prediction. As shown, even the leading teams achieved limited success, around 75 points, while most teams scored below 25 points, indicating low accuracy. These results demonstrate the considerable challenge of accurately predicting protein structures at the time.
Google DeepMind's AlphaFold made its debut four years later, in 2018, during the CASP13 competition. The results graph indicates that AlphaFold's prediction accuracy was markedly superior, at a level completely different from before.
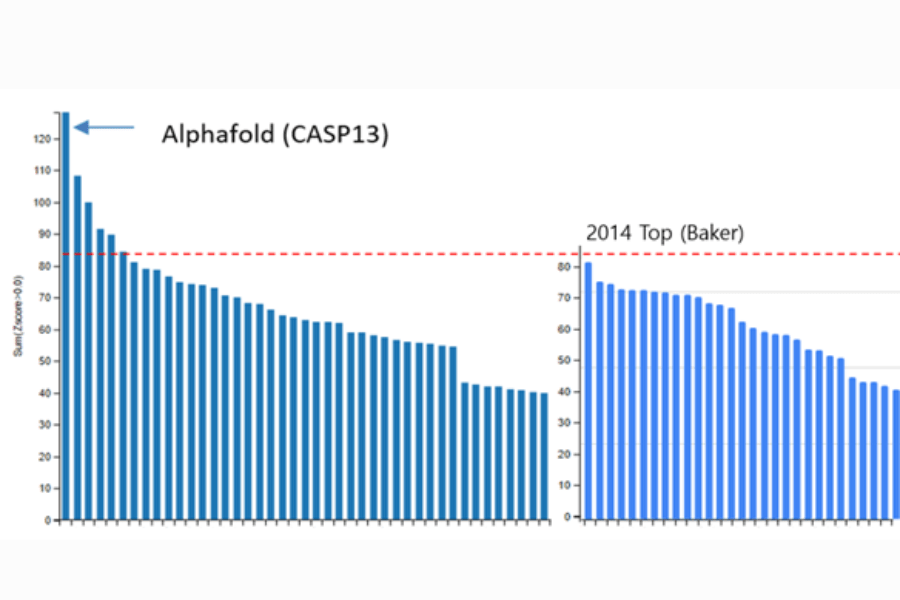
Comparing its performance to the top team from four years earlier illustrates the progress made. While Baker's team scored a maximum of around 80 points in 2014, AlphaFold achieved an accuracy close to 120 points in 2018. This signifies a substantial leap in the field of protein structure prediction.
How could AlphaFold achieve such remarkable results? Everyone at the competition waited for the AlphaFold team to reveal their experimental model, and the AlphaFold team explained their approach this way (Senior et al., 2020).
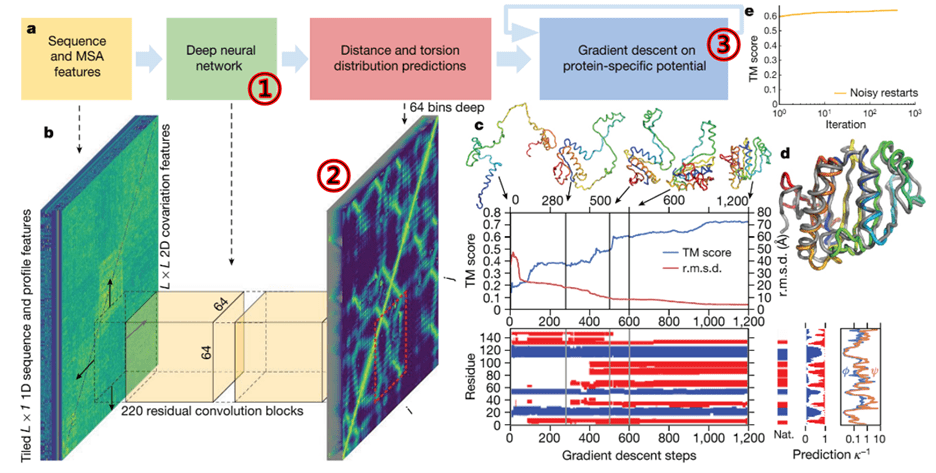
It may seem complex, but the process involves using CNNs (as covered in the first column) within a deep neural network. Since CNN technology identifies important features through image analysis, the diagram illustrates how they transformed 3D structural information into 2D feature maps for analysis. The process also employed commonly used gradient descent methods for optimization.
Protein structure prediction researchers began to understand how AlphaFold achieved such impressive results. Each began preparing new challenges, incorporating their own ideas, with hopes of surpassing AlphaFold in the next competition. For example, regarding the Baker team's work: The principle AlphaFold used to convert distances into a 2D image is illustrated here:

This involved measuring distances between amino acids (specifically, C-alpha atoms) and transforming this into a 2D image representation. Observing this, the traditional research teams that had been participating in CASP while AlphaFold effectively utilized distances and how the backbone bends, it might not have fully leveraged information about the orientations between residues facing each other. Consequently, they developed new models that incorporate this additional features.

As the graph shows, some of the tools developed by the traditional CASP participating teams surpassed AlphaFold's performance. (Yang et al., 2020).
Naturally, everyone eagerly awaited the results of the upcoming competition. Would the traditional teams catch up to AlphaFold?
Here are the results:

As the graph illustrates, it was a landslide victory for the Google DeepMind team, which introduced a new model called AlphaFold2. They scored close to 240 points, while all other teams, including Baker's, remained around 90 points. Comparing this result to previous ones is even more astonishing. It was a groundbreaking achievement, far surpassing not only the Baker team's top score of around 80 from six years prior but also their own AlphaFold1 score of nearly 120 from two years earlier. For creating AlphaFold2, Hassabis and Jumper, along with Baker — the long-time expert in the field, were jointly awarded this year's Nobel Prize in Chemistry.
So, how did AlphaFold2 manage to not only outperform all previous attempts by a wide margin but also significantly surpass its predecessor, AlphaFold1? This can be viewed from two perspectives (Jumper et al., 2021).

The first point to note is that they moved beyond the constraint of using only predetermined distance information, such as C-alpha distances, and utilized the sequence information itself, including MSA and pair representation, as indicated in the input stage of the diagram. Relying solely on known distances requires confirmed structural information, much like needing the finished balloon art to replicate it. However, learning from the sequence itself means breaking the convention of predicting based on finished structures and enabling learning even from just the unfolded sequence, similar to the long balloon.
The second point to note is the module labeled 'Evoformer' in the diagram. This is a modification of the Transformer algorithm. The Transformer is the algorithm that powers today's ChatGPT and is a very powerful tool for understanding sequence characteristics (Vaswani et al., 2017). Essentially, advancements in deep learning algorithms, particularly attention mechanisms like those in Transformers/Evoformers, have elevated AI models to a point where they can learn complex relationships directly from sequences.
To summarize again, the emergence of AlphaFold1 and the subsequent development of AlphaFold2 demonstrate the rapid evolution of AI technology. Furthermore, it can also be stated that AI has far surpassed the pace of researchers operating within traditional academic constraints.
There's a critical point here. Just as researchers who have long studied protein structure prediction have struggled to keep up with the pace of AI development, AI has advanced at an astonishing rate, propelled by decades of accumulated data. Recently, it has reached a level where it can independently deduce principles from given information. This means that in the future, many more scientific challenges may see breakthroughs through AI.
This phenomenon presents us with important considerations. As the pace of AI technology development accelerates, we must thoughtfully evaluate how we will utilize this technology and what role we should assume amidst these changes. The next column will examine the current situation in detail, caused by the rapid development of AI, and discuss potential response strategies.
References:
1. Jo, T., Hou, J., Eickholt, J., & Cheng, J. (2015). Improving protein fold recognition by deep learning networks. Scientific Reports, 5, 17573.
2. Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., ... & Polosukhin, I. (2017). Attention is all you need. Advances in Neural Information Processing Systems, 30.
3. Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., ... & Hassabis, D. (2020). Improved protein structure prediction using potentials from deep learning. Nature, 577, 706-710.
4. Yang, J., Anishchenko, I., Park, H., Peng, Z., Ovchinnikov, S., & Baker, D. (2020). Improved protein structure prediction using predicted interresidue orientations. Proceedings of the National Academy of Sciences, 117(3), 1496-1503.
5. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., ... & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583-589.